How to call whole genome MLST on the command line
I have been asked in many different contexts how to call alleles on the command line. In August 2022, I gave an internal workshop on how to call alleles on the command line and I thought I would translate that into a blog post.
If you’re here to look at classic MLST callers, this isn’t that blog post.
If you want to know more about classic MLST callers, please see our paper:
Page et al 2017, “Comparison of classical multi-locus sequence typing software for next-generation sequencing data.” Microbial Genomics
NOTE: I will give the caveat that ChewBBACA has gone from version 2 to version 3 and so it is possible that things have changed.
Summary
The three callers that seem to be great on the command line are
Each caller has its positives and negatives and so I cannot say that one or the other is clearly the best one over all others.
A lot of my motivation is to find a CLI supplement for BioNumerics’s wgMLST caller, and so I compared results against BioNumerics. I had received four benchmark datasets from PulseNet and made a scatterplot of MLST distances between any two genomes in BioNumerics and the other caller. The distances used in the scatterplot are number of identical alleles divided by the number of total loci compared. Missing loci were excluded from the denominator.
When looking at the trendline between callers and BioNumerics, the linear relationship is outstanding.
| Dataset | Caller | Trendline | R2 |
|---|---|---|---|
| Salmonella enterica | ChewBBACA | Y = 1.01x - 0.01 |
1 |
| Salmonella enterica | EToKi | Y = 1x + 0 |
1 |
| Salmonella enterica | ColorID | Y = 1.01x - 0.01 |
1 |
| STEC | ChewBBACA | Y = 1x - 0.01 |
1 |
| STEC | EToKi | Y = 0.99x + 0 |
1 |
| STEC | ColorID | Y = 0.99x + 0 |
1 |
| Campylobacter jejuni | ChewBBACA | Y = 1x + 0 |
1 |
| Campylobacter jejuni | EToKi | Y = 1x + 0 |
1 |
| Campylobacter jejuni | ColorID | Y = 1.01x + 0 |
1 |
| Listeria monocytogenes | ChewBBACA | Y = 1x + 0 |
1 |
| Listeria monocytogenes | EToKi | Y = 1x + 0 |
1 |
| Listeria monocytogenes | ColorID | Y = 1x + 0 |
1 |
I also took a random Campylobacter jejuni genome and ran the same genome assembly through each of the three callers 50 times each. Even if the caller had the option for multithreading, I kept it at 1 thread each.
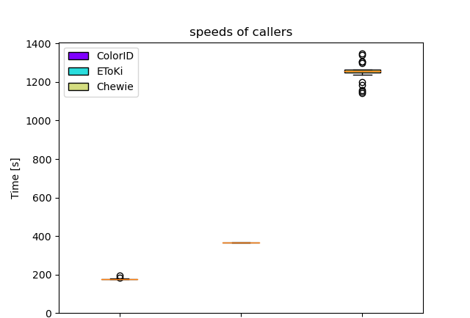
This table is my semi-subjective overall review of the callers.
| Measure | EToKi | ChewBBACA | ColorID |
|---|---|---|---|
| Algorithm | Gene prediction, “uberblast” to ref alleles to get locus, then hashsum comparison against complete database for allele call. | Gene prediction, BLASTp to ref alleles to get locus, then BLASTp to complete database for allele call. | Colored Debruijn graphs |
| Developers | Single developer; Slightly responsive | PhD students; responsive | Single developer; Very responsive |
| Documentation | Minimal | Extensive wiki | Medium |
| Installation | Source; (Conda); Docker | Source; Conda; Docker | Cargo (easy) |
| Adding new alleles | Not yet; should be easy to add | Automatically done but also alters the database without approval | Cannot be done but could be a new feature |
Let’s call alleles
We’ll turn this overview into a workshop starting here.
Get your data
- Make a folder to stay organized. The code for this list is below this list.
- Get a database from https://chewbbaca.online. For this workshop, I am going to use https://chewbbaca.online/api/NS/api/download/compressed_schemas/4/1/2021-05-30T22:06:50.902917 which is a download link all the way to the right side on the Campylobacter page.
- Get an assembly you want to call alleles with. I chose 15AR0919 at random for this blog post since it is a complete genome.
mkdir -pv ~/workshops/wgMLST
cd ~/workshops/wgMLST
mkdir Campy.chewbbaca
cd Campy.chewbbaca
wget -O Campylobacter_jejuni_INNUENDO_wgMLST_2021-05-30T22_06_50.902917.zip "https://chewbbaca.online/api/NS/api/download/compressed_schemas/4/1/2021-05-30T22 :06:50.902917"
unzip Campylobacter_jejuni_INNUENDO_wgMLST_2021-05-30T22_06_50.902917.zip
cd ..
mkdir asm
# I took this download link from interacting on the NCBI website at https://www.ncbi.nlm.nih.gov/nuccore/NZ_CP035894.1
wget -O asm/15AR0919.fasta "https://www.ncbi.nlm.nih.gov/sviewer/viewer.cgi?tool=portal&save=file&log$=seqview&db=nuccore&report=fasta&id=1804576668&&ncbi_phid=null"
What to expect when calling alleles
For each caller, here is what you should expect:
- Will take a few minutes on a bacterial genome
- Output formats
- tsv profiles file
- Assembly name, usually in the first column
- Allele integers, usually in subsequent columns
- fasta of allele matches (not always)
- tsv profiles file
ChewBBACA
https://github.com/B-UMMI/chewBBACA
ChewBBACA Installation
ChewBBACA can be installed via conda
conda create -c bioconda -c conda-forge -n chewie "chewbbaca=3.1.2"
conda activate chewie
ChewBBACA Calling alleles
Calling alleles is pretty easy and takes about 2 minutes with 8 cpus. It is efficient in batching and so your times might speed up per genome if you are able to query multiples at once.
chewie AlleleCall -i asm --schema-directory Campy.chewbbaca -o chewie.out --cpu 8
For chewie version 3, you can add --no-inferred to avoid adding new alleles to the database as you are querying.
For chewie version 2, you can add --fr to ignore any temporary files in the database and start fresh.
ChewBBACA Results
$ ls chewie.out/
cds_coordinates.tsv loci_summary_stats.tsv paralogous_counts.tsv results_alleles.tsv results_statistics.tsv
invalid_cds.txt logging_info.txt paralogous_loci.tsv results_contigsInfo.tsv
Summary results are valuable and so let’s look at those first. The really great thing is that each of these columns are documented very well and are well-thought out.
$ column -t results_statistics.tsv
FILE EXC INF PLOT3 PLOT5 LOTSC NIPH NIPHEM ALM ASM PAMA LNF
15AR0919 1078 68 0 0 0 2 4 0 0 0 1642
Here, it looks like we have 1078 alleles that match the Campylobacter wgMLST scheme exactly and then 68 inferred (new) alleles. 1642 loci were not found but that is not a big deal since we always expect many accessory loci not to be found. Actually, it would be awful if we found all loci in a single genome because it would show that something really weird was happening.
The other really important file is results_alleles.tsv which shows all allele integers per locus.
$ cut -f 1-5 results_alleles.tsv | column -t
FILE INNUENDO_wgMLST-00013230 INNUENDO_wgMLST-00013231 INNUENDO_wgMLST-00013232 INNUENDO_wgMLST-00013233
15AR0919 LNF 27 1 14
Here, I am showing the first five columns where the first column is the assembly name,
and then the next columns are loci and their alleles.
The first locus was not found as indicated by LNF.
The next loci were found to be alleles 27, 1, and 14.
EToKi
EToKi is the MLST caller behind EnteroBase and is at https://github.com/zheminzhou/EToKi. Unfortunately, the developer has not responded to any github issues or pull requests recently. However, I found some issues: not all processes had 1 CPU by default. Not all processes had the same default CPU. Additionally, it depended on a license-restricted software package USEARCH. Therefore, I fixed it into my fork at https://github.com/lskatz/EToKi. Then, @rpetit3 helped us get it into Conda based on my fork.
EToKi installation
conda install -n etoki etoki
EToKi format database
As opposed to ChewBBACA, you cannot download the EToKi-formatted database directly You need to reformat the ChewBBACA database. I used seqtk and perl one-liners to do so.
This section makes a full concatenation on all fasta files from the Chewie database while making a two-line-per-entry fasta file (id, sequence).
mkdir -pv ~/workshops/wgMLST/Campy.etoki
cd ~/workshops/wgMLST
cat Campy.chewbbaca/*.fasta | \
seqtk seq -l 0 - > Campy.etoki/source.fasta
This section removes new alleles indicated by * and then removes problematic _ characters.
EToKi uses _ as a reserved character in separating locus and allele in a defline.
cd Campy.etoki
cat source.fasta | \
perl -lane '
$id=$_; $seq=<>; chomp($id,$seq); $id=~s/INNUENDO_|Pasteur_//; next if($id=~/\*/); $underscores = () = $id=~/_/g; die "ERROR: $id" if($underscores > 1); print "$id\n$seq";
' > etoki.fasta
Next for EToKi, we need to make a references file: a file of one allele per locus. Usually, the first allele in an MLST fasta file is the representative allele and therefore the one we are taking.
cat etoki.fasta | \
perl -lane '
$id=$_; $seq=<>; chomp($id,$seq); ($locus,$allele) = split(/_/, $id); next if($seen{$locus}++); print "$id\n$seq";
' > etoki.refs.fasta
cd ..
Next, use EToKi to create an md5sums database file for the rest of the alleles.
This file will be etoki.csv.
EToKi.py MLSTdb -i Campy.etoki/etoki.fasta -r Campy.etoki/etoki.refs.fasta -d Campy.etoki/etoki.csv
EToKi Calling Alleles
Run EToKi’s caller like so
EToKi.py MLSType -i asm/15AR0919.fasta -r Campy.etoki/etoki.refs.fasta -k 15AR0919 -d Campy.etoki/etoki.csv -o 15AR0919.etoki.fasta
EToKi uses the references fasta file (-r) to find the correct locus and then the csv database (-d) to see if it is an exact match to any existing allele.
If it is close enough to the reference but not an exact match to any known allele,
it will indicate it in the resulting fasta file.
-k will simply apply a sample name to the results.
The results file is a fasta file with a detailed defline for each entry
and the allele sequence.
EToKi Results
There is a resulting fasta file 15AR0919.fasta that contains each allele
in a fasta entry.
The defline is very detailed and so here is one example.
grep -m 1 ">" 15AR0919.fasta
>wgMLST-00013231 value_md5=13777d1c-21fb-e85c-1678-e85bf2a9a586 id=27 CIGAR=wgMLST-00013231_1:648M accepted=2 reference=MLSType:15AR0919 identity=0.9906999999999999 coordinates=NZ_CP035894.1:1124613..1125260:+
The fields can be briefly explained like
>wgMLST-00013231locus namevalue_md5=md5sum of sequenceid=1allele 1 of this locusCIGAR=A cigar string describing the matchaccepted=2a bitwise flag describing the match. 1 or 2 indicate good matches1a good allele2an allele that has no major problem in its sequences, but may be low quality (still acceptable).2is used by default when there is no quality scores such as a fasta file input.8the allele is fine, but we are not the central database, so will not be able to assign a formal ID32the gene is duplicated (two or more hits in the genome)64the gene is fragmented256the identity to the reference is too low
reference=The etoki module MLSType and the sample nameidentity=0.99...99% matchcoordinates=query assembly coordinates
EToKi: Extra step to make the profiles spreadsheet
Next, we need to turn this output fasta file into a profiles file, which is a spreadsheet of alleles. To do that, you can use my custom perl script.
pushd $HOME/bin
git clone git@github.com:lskatz/lskScripts.git
export PATH=$PATH:$HOME/bin/lskScripts/scripts
The usage for this script can be pulled up with matrix.etoki.pl --help
matrix.etoki.pl: Generate a matrix of alleles from a set of etoki fasta results
Usage: matrix.etoki.pl [options] *.etoki.fasta > matrix.tsv
where etoki.fasta files are EToKi results with MLST deflines
OPTIONS
--database The path to the etoki database whose format is CSV with
three columns: md5sum, locus, allele [required]
--help This useful help menu
And then from there, it’s just one command to make the EToKi spreadsheet
matrix.etoki.pl --database Campylobacter_jejuni.etoki/etoki.csv *.etoki.fasta > etoki.tsv
ColorID
ColorID is based on a newer algorithm called colored de bruijn graphs. The database creation is intense but querying on that database is incredibly fast. The major downside of ColorID vs other algorithms is that it cannot report new alleles.
ColorID Installation
ColorID is programmed in Rust and therefore you can install it with cargo.
pushd $HOME/bin
git clone https://github.com/hcdenbakker/colorid.git
cd colorid
cargo build --release
export PATH=$PATH:~/bin/colorid/target/release:~/bin/colorid/scripts
popd
Add the export PATH command into .$HOME/.bashrc to keep the new path permanent.
ColorID Format Database
For the version of MLST with Color ID, we actually compare the MLST database against the query. Therefore, the assembly or the raw reads get formatted.
mkdir colorid.out
bxi="colorid.out/assembly.bxi"
fofn="chewie.out/asm.fofn
# We currently only have one query fasta file in this example
# but it is worth showing how it would look like with any number of fasta assembly files.
\ls asm/*.fasta | xargs -P 1 -n 1 bash -c 'echo -e "$(basename $0 .fasta)\t$0"' > $fofn
colorid build -b $bxi -s 30000000 -n 2 -k 39 -t 4 -r $fofn
The ColorId build command:
-b $bxiis where the index is going to be saved-s 30000000is the size of the bloom filter. You want it to be about 10x the size of the genome or greater. Campy is about 1.6M and so 30M is fine.-n 2is the number of hashes to use for the the bloom filter-k 39is the kmer size. 39 seems to work well with bacteria.-t 4is 4 threads-r $fofnis the file of filenames of assemblies to put into the database.
It should look something like this after the colorid build command.
Ref_file : colorid.out/asm.fofn
Bigsi file : colorid.out/assembly.bxi
K-mer size: 39
Bloom filter parameters: num hashes 2, filter size 30000000
Inference of Bloom filters in parallel using 4 threads.
Creation of index, this may take a while!
Saving BIGSI to file.
And therefore your database is the query fasta file. In the next step, your actual query will be the MLST database against your assemblies.
ColorID Calling Alleles
In the query step, we match the MLST database against the fasta assembly file(s).
We can simply use the ChewBBACA database as-is since it is a folder of fasta files.
We use -s to indicate that we only want perfect matches.
We use -m to indicate that each accession in the MLST database will be treated as a separate query.
colorid search -ms -b $bxi.bxi -q Campylobacter_jejuni.chewbbaca/*.fasta > colorid.out/alleles.colorid.txt 2> colorid.out/alleles.colorid.log
# Delete any line with custom alleles as indicated with a * in the identifier
$ sed -i.bak '/\*/d' colorid.out/alleles.colorid.txt
Next, we want to transform these raw results into a profile spreadsheet of alleles.
ColorID has a python script process_MLST.py to do that.
# Transform to spreadsheet
process_MLST.py colorid.out/alleles.colorid.txt colorid.out/mlst
cut -f 1-4 colorid.out/mlst.detailed.tsv | column -t
# Gives a profiles spreadsheet of alleles.
Turning alleles into distances
After each of the MLST calling steps, you should have a spreadsheet of alleles. Typically, this is tab delimited with the first column as the identifier of the sample. The subsequent columns are labeled with each locus and the values are the alleles. In most cases, these allele values are simple integers; in other cases, they could be hashsums of allele sequences.
Great, now what? 🤷
Now we need to turn these alleles into distances. These distances can either be in the form of a distance matrix or in the form of a tree.
Here are the facts going into it.
- We have a tsv for any caller in the same format.
- One row is a header
- Other rows are the sample name, followed by integers.
- There are some differences between callers in their profiles spreadsheet.
- Missing alleles are indicated differently.
- BioNumerics:
? - EToKi:
-1(this nuance is due to my custom perl script and not the original author) - ChewBBACA:
LNF - ColorID:
MULTIorNOT_CALLED
- BioNumerics:
- The sample name might be named differently. Maybe the full fasta filen ame or maybe the basename of the filename.
- Missing alleles are indicated differently.
I have created perl scripts for each caller to help identify distances. Additionally, we have found that GrapeTree is an excellent method already in place to generate trees from an allele profile spreadsheet.
ChewBBACA distances
distance matrix
distance.chewbbaca.pl chewie.out/results_alleles.tsv > chewie.distances.tsv
tree
grapetree --profile chewie.distances.tsv
EToKi distances
distance matrix
distance.etoki.pl *.etoki.fasta > etoki.distances.tsv
tree
grapetree --profile etoki.distances.tsv
ColorID distances
distance matrix
distanec.coloridmlst.pl colorid.out/alleles.colorid.txt > colorid.distances.tsv
tree
grapetree --profile colorid.distances.tsv
Done!
I went through a lot of background and hands on instruction and so by this point you should have a basic knowledge of
- cgMLST methods
- How to format cgMLST DBs
- How to call alleles
- Finding distances with alleles
- Creating trees with grapetree
Appendix
Callers excluded from the workshop/study
| Caller | Intended use | Reason for exclusion |
|---|---|---|
| mlst | 7-gene MLST | Did not call enough loci |
| ARIBA | Antimicrobial resistance identification By Assembly (and often, 7-gene MLST) | Too slow for a full genome |
| SRST2 | 7-gene MLST via short reads | Too slow for a full genome |
| Mentalist | wgMLST | Dependencies are broken and the developer is unresponsive. I could not install it. |
| BigsDB | wgMLST and 7-gene MLST | Could not separate the server from the allele caller |
| StringMLST | 7-gene MLST | Did not call enough loci |
| BioNumerics | wgMLST | Sunsetting |
| SeqSphere+ | wgMLST | License restricted |
| PyMLST | wgMLST | Batch queries not possible |
Callers not examined
| Caller | Intended use | Why not examined |
|---|---|---|
| KMA-cgMLST | cgMLST | I was not aware of it at the time of this workshop/study |
| STing | 7-gene MLST | Likely the same issue as StringMLST since it is a similar algorithm |